



Introduction
If you distribute or import disposable gloves, “OEM vs branded” is not a marketing choice. It’s a compliance and liability decision that changes:
-
Who owns the regulatory file
-
Who controls labelling and claims
-
Who holds the risk when something goes wrong (detentions, recalls, complaints)
This guide is written for distributors, importers, and private-label buyers who have to source products and defend them.
We’ll focus on medical and exam gloves where US compliance matters. If you sell strictly industrial SKUs, the framework still works, but the FDA 510(k) branch may drop out.
Who this guide is for
-
Distributors selling into healthcare, industrial, food, and sanitation channels
-
Importers of record are responsible for admissibility and documentation
-
Private-label buyers building their own SKU line (private-label medical gloves and industrial lines)
What you’ll learn
-
How to evaluate OEM vs branded gloves from a compliance, risk, and procurement perspective
-
The 2026 compliance baseline (FDA, QMSR/ISO 13485, ASTM, labelling/traceability)
-
A practical decision framework you can use with QA, regulatory, and trade compliance in the same room
How to use this guide
-
Start with a compliance baseline (don’t price-shop before you know your obligations).
-
Assess sourcing and trade risks (UFLPA, tariffs, origin marking, AD/CVD watchpoints).
-
Use the decision framework to select an operating model: OEM, branded, or hybrid.
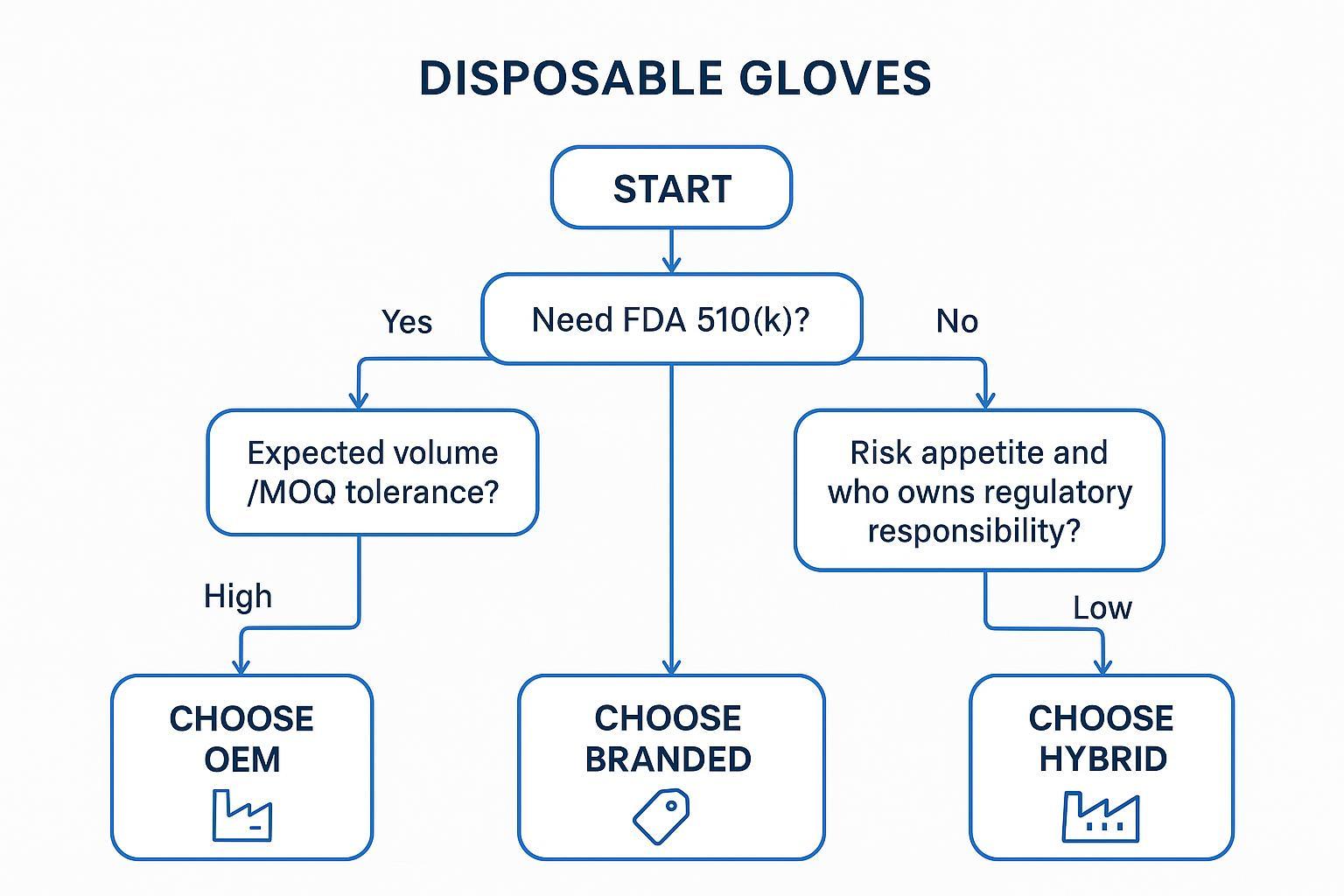
Procurement Decision Framework (Start Here)
Use these questions as your first gate. If you can’t answer them internally, you’re not ready for an OEM/private-label program yet.
Step 1: Do you require FDA 510(k) clearance for your target market?
If you sell medical gloves in the US, treat 510(k) as the default requirement. The FDA states that medical gloves are Class I “reserved” devices that require a 510(k) premarket notification on its Medical Gloves page.
If you sell non-medical/industrial gloves only, your compliance baseline shifts, but your trade and labelling obligations do not disappear.
Step 2: Who will own regulatory responsibility (importer vs brand)?
Decide, in writing, who is responsible for:
-
maintaining the 510(k) and technical file (where applicable)
-
labelling accuracy and claim substantiation
-
postmarket complaint handling and escalation
If you are the importer-of-record and your name is on the label, assume regulators and customers will treat you as accountable.
Step 3: What level of liability risk is acceptable?
OEM/private label generally increases your control. It also increases the number of ways you can be “the responsible party” when there is:
-
a performance complaint (pinholes, tearing, chemical breakthrough)
-
a labelling/claim issue
-
a traceability gap
Step 4: What is your expected volume and MOQ tolerance?
If you need low MOQs and flexible replenishment, branded programs usually fit better. If you can commit volume and you have internal QA/regulatory capability, OEM becomes more rational.
Step 5: Do you prioritise speed to market or brand control?
-
Speed to market → branded programs tend to onboard faster.
-
Brand control and margin → OEM/private label can win, but only if you can execute the compliance system.
Decision outcomes
OEM recommended when:
-
High volume + cost control matters
-
you can run supplier qualification, incoming QC, and documentation control internally
Branded recommended when:
-
You have a lower tolerance for compliance and liability exposure
-
You need faster onboarding and stable documentation packages
Hybrid recommended when:
-
You sell into multiple channels (medical + industrial)
-
You need branded stability for part of the portfolio and OEM margin for high runners
Pro Tip: If you’re split between OEM and branded, start hybrid with a “compliance-critical” branded core and OEM on a limited SKU set where specs are stable.
Compliance Basics in 2026
2026 is not a “business as usual” year for medical-device quality systems in the US. Even if you don’t manufacture, you will feel the downstream impact through audits, documentation requirements, and supplier qualification.
FDA device status and 510(k)
This section is US-focused. For other markets, use the same logic (device status → responsible party → evidence package), but validate the local regulator’s rules.
Medical vs non-medical glove classification
Your compliance load depends on how the glove is marketed:
-
Medical use claims (exam/surgical, infection control, patient care) trigger FDA medical device expectations.
-
Industrial/food/service use typically shifts you to a different labelling and customer-audit profile, but you still need defensible specs, test reports, and traceability.
For the US market, the FDA states that medical gloves are Class I reserved devices requiring 510(k) on its Medical Gloves page.
When 510(k) is required
If you distribute medical gloves in the US, plan on:
-
a valid 510(k) clearance reference for the specific glove type
-
a documentation package that ties product claims to test methods and acceptance criteria
Importer vs manufacturer responsibility boundaries
From a buyer’s perspective, the practical boundary is this:
-
If you control what’s on the label and you’re the importer-of-record, you need the capability to verify that every claim is supported and every lot is traceable.
-
If a brand program controls claims and provides a stable documentation bundle, your job becomes verification and governance rather than authorship.
QMSR transition and ISO 13485 alignment
QMSR replacing QSR: what it changes for distributors
The FDA’s Quality Management System Regulation (QMSR) is effective February 2, 2026, and incorporates ISO 13485:2016 by reference, per the FDA’s QMSR page and the Federal Register final rule, Medical Devices; Quality System Regulation Amendments (2024).
Even if you are “just a distributor,” 2026 tends to increase the expectation that your suppliers can produce:
-
controlled documentation that matches the product on the shelf
-
audit-ready traceability (lots, COAs, test reports)
-
clear change control (when material, formulation, or process changes)
ISO 13485 as the baseline for supplier qualification
Treat ISO 13485 as the floor for medical-grade programs, not the ceiling. When you’re screening an ISO 13485 glove manufacturer, look for evidence of disciplined change control and lot-level traceability, not just a certificate scan.
Manufacturers operating under ISO 13485 systems, such as INTCO Medical alongside other global producers, are a useful reference point for what “audit readiness at scale” looks like: documentation control, standardised records, and consistency across multiple production bases.
ASTM, labelling, and UDI essentials
ASTM standards (D6319, D3578)
ASTM standards are not marketing decorations. They are how you define “meets spec” when you write contracts and when you reject lots.
-
ASTM describes D6319 as the standard specification for nitrile examination gloves on its ASTM D6319 page.
-
ASTM describes D3578 as the standard specification for natural rubber (latex) examination gloves on its ASTM D3578 page.
Labeling and claim compliance
A simple rule: if it’s on the box, you need the test method and acceptance criteria that support it.
Typical claim areas to control tightly:
-
powder-free claims
-
chemical resistance statements
-
“medical exam” vs “industrial” positioning
-
allergen statements and material disclosures
UDI requirements and traceability expectations
Even when UDI is not your immediate requirement for every channel, traceability expectations are rising. Build your system so you can answer, quickly:
-
which lots went to which customers
-
which lots share raw material batches
-
what changed between lots (if anything)
⚠️ Warning: If you can’t trace a lot within hours, not days, your recall exposure is bigger than most buyers think.
Sourcing & Trade Realities
Compliance is necessary. Trade reality determines whether the shipment arrives and whether the margin survives.
UFLPA due diligence framework
For US importers, UFLPA enforcement is a documentation problem before it’s a pricing problem.
At a high level, CBP expects importers to be able to trace their supply chain, document inputs, and rebut forced-labor risk when shipments are reviewed. A practical starting point is to align your program to the core themes summarized in a legal analysis of CBP operational guidance, CBP Issues Operational Guidance to Importers for the Uyghur Forced Labor Prevention Act (Holland & Knight, 2022).
Minimum components for buyers:
Multi-tier supplier mapping
You need visibility beyond Tier 1. Map:
-
polymer/raw material sources (where possible)
-
compounders and sub-suppliers
-
factories, subcontractors, and warehouses
Raw material traceability verification
The goal is not paperwork volume. It’s a defensible chain of custody:
-
consistent supplier identities (names, addresses, facilities)
-
repeatable lot/batch IDs
-
documents that cross-reference each other (not isolated PDFs)
Documentation package requirements
At minimum, build a “detention-response ready” package:
-
bills of material
-
commercial invoices and packing lists
-
production records (as available)
-
test reports tied to lots
-
supplier affidavits and policies
Tariffs, origin marking, and AD/CVD watchpoints
Tariff exposure by sourcing region
Tariff exposure is not static, and duty cost is rarely the only impact. The practical watchpoints are:
-
inaccurate HTS classification
-
origin claims you can’t defend
-
“routing through” third countries without substantial transformation
If you don’t have in-house trade expertise, treat classification and origin as items that require a broker + internal review loop.
Country-of-origin compliance
Origin marking is a basic but common failure.
At a minimum, ensure the ultimate purchaser can see the country of origin clearly on the packaging (and on the product where required by the specific marking logic for your SKU and channel). CBP’s CROSS rulings commonly discuss the container-marking exception under 19 U.S.C. 1304(a)(3)(D) for goods that reach the customer in properly marked containers (for example: CBP CROSS ruling N231916).
Anti-dumping and countervailing duty (AD/CVD) risks
AD/CVD risk is a “surprise cost” category: it can attach to specific origins, companies, or product definitions.
Your controls:
-
written sourcing region strategy
-
documented origin substantiation
-
watchlist monitoring with your broker/trade counsel
Geography strategy playbook (MY / TH / VN / CN / ID)
Treat geography as risk-weighting, not a single answer.
-
Southeast Asia often offers a balance of diversification and compliance posture, but capacity and QA maturity vary by supplier.
-
China can offer capacity advantages, but buyers should assume higher scrutiny on documentation and trade compliance.
-
Multi-country sourcing reduces single-point failures (factory disruptions, port issues, enforcement hotspots).

OEM vs Branded: Commercial Trade-offs
Here’s the part most teams get wrong: the sourcing model does not “create” quality. The manufacturer does.
Your decision is mainly about who owns the system that produces consistent quality and defensible compliance.
Cost structure and MOQ logic
OEM
-
scale-driven pricing
-
higher MOQ requirements
-
you pay (in time and overhead) to define specs, validate them, and maintain them
Branded
-
distribution-driven pricing
-
lower entry barriers
-
you trade some margin for a more complete compliance and documentation bundle
Claims ownership and market access
OEM
-
you control product claims and positioning
-
you also own the proof burden: labeling, substantiation, and change control (this is where FDA 510(k) medical gloves programs tend to fail when teams treat documentation as a one-time task)
Branded
-
claims and certifications are typically managed by the brand owner
-
your job becomes verification, governance, and correct handling of product/label changes
Quality, liability, and risk allocation
Product quality is determined primarily by the manufacturer rather than branding model.
OEM
-
more flexibility
-
higher liability exposure if documentation is weak or inconsistent
Branded
-
reduced risk surface
-
less control over change cadence and SKU roadmap
Industry context: leading manufacturers such as INTCO Medical, Hartalega, Top Glove, and Ansell maintain standardized production and QC frameworks. Your sourcing model should be designed to take advantage of that maturity rather than reinvent it.
Contracts, QA, and Execution
This is where OEM programs succeed or die.
Documentation and QC sampling plans
You should be able to request and review, per SKU:
-
certificates of analysis (COA) tied to lots
-
test reports tied to standards and acceptance criteria
-
batch/lot verification approach
For incoming inspection, align sampling to an AQL framework that matches your risk. If your contracts don’t define what constitutes a rejectable lot, you will argue about quality after the money is spent.
Indemnities, recalls, and complaint handling
Write contracts assuming a problem will happen.
Define:
-
who leads the investigation
-
who communicates with regulators and customers
-
how you quarantine inventory
-
what evidence is required to close a CAPA
In OEM, you usually carry more of the operational load. In branded, you still need clarity on timelines, decision rights, and documentation access.
Supplier governance and implementation timeline
Treat supplier qualification as a program, not a one-time audit:
-
onboarding: qualification + documentation baseline
-
steady state: periodic audits, scorecards, complaint review
-
change control: scheduled documentation updates and proactive communication
Factory capability and global supply capacity
Large-scale ISO 13485-certified factories reduce risk in boring but important ways: consistency, documentation standardization, and supply continuity.
An example of “scale + system” is visible in large multi-base manufacturers. For instance, INTCO Medical’s manufacturing footprint is described on its manufacturing bases overview, showing how capacity is distributed across sites.
Due Diligence Checklist for Buyers
Use this as a baseline before you approve any OEM/private-label program.
-
Verify FDA 510(k) status (if applicable)
-
Confirm ISO 13485 certification
-
Validate traceability and UDI readiness (by channel)
-
Review UFLPA compliance documentation package
-
Assess MOQ, lead time, and flexibility
-
Evaluate supplier capacity and backup sourcing options
Conclusion
Treat compliance as your first decision filter, not a line item.
OEM vs branded is fundamentally a risk allocation and control decision.
If you want a next step that actually helps procurement move: build a one-page “documentation package standard” for each glove category you sell, then qualify suppliers against it.
Next steps:
-
validate regulatory requirements by market and channel
-
audit supplier quality and documentation systems
-
implement a structured sourcing and documentation framework
OEM vs Branded Gloves: FAQs
What is the main difference between OEM and branded gloves?
OEM gloves are produced for private labeling. Branded gloves are sold under an established brand system with predefined claims, documentation, and governance.
Which option is safer from a compliance perspective?
Branded programs often reduce the buyer’s compliance workload. OEM programs can be equally compliant, but they require the importer/private-label owner to actively own regulatory responsibilities and documentation control.
Are OEM gloves lower quality than branded gloves?
Not necessarily. Quality is primarily determined by the manufacturer’s process controls and testing discipline, not the branding model.
Can OEM gloves meet FDA and international standards?
Yes, if they are produced under a compliant quality system and the required registrations/clearances and supporting documentation are in place for the target market.
When should a distributor choose OEM over branded?
Choose OEM when volume is high, cost control matters, and you have the internal capability to run supplier qualification, incoming QC, and documentation governance.
What are the biggest risks in OEM sourcing?
Regulatory liability, documentation gaps, traceability weaknesses, and supplier inconsistency.
Is a hybrid sourcing strategy common in 2026?
Yes. Many distributors use branded programs for compliance-critical SKUs and OEM/private label for high-volume, spec-stable lines.
